Patern Distrofiler
Okuma:2886
Retina pigment epitelinin (RPE) patern distrofisi (PD), makülada ve çevresinde gri veya sarı renkli pigment birikimi ile karakterize nadir görülen fundus değişiklikleridir. Bu pigment depozitleri farklı noktalar, çizgiler, dallanmalar şeklinde veya retiküler şekilde olabilir. Klasik patern distrofi biçimi, Deutman ve ark. tarafından 1970'de tanımlanmış olan kelebek şekilli distrofidir (KD). Daha sonra Gass, pigment dağılımının biçimine göre patern distrofiyi 5 temel distrofi grubuna ayırmıştır: Erişkin başlangıçlı (Adult) foveomaküler vitelliform distrofi (EBFVD), fundus pulverulentus (FP), fundus flavimakülatusu taklit eden multifokal PD (FFpD), kelebek şekilli distrofi (KD) ve retiküler distrofi (RD). Çakışan özellikleri ve benzer klinik seyirleri nedeniyle, pek çok yazar PD genel terimini yeterli bulmaktadır. Bazı yazarlar ise, PD terimini yalnızca kelebek şekilli distrofi için kullanmakta ve retiküler distrofiyi ayrı bir RPE distrofisi olarak kabul etmektedir. Ayrıca, erişkin başlangıçlı foveomaküler vitelliform distrofi bazen Best vitelliform distrofinin polimorf şekli olarak da kabul edilmektedir. Altıncı kromozomun kısa kolundaki RDS/periferin genindeki pek çok mutasyonun PD ile ilişkili olduğu gösterilmiştir.
Patern distrofi, spesifik semptomunun olmayışı, ileri yaşlara kadar oldukça iyi santral görme düzeyinin korunması, normal renkli görme ve görme alanı ile karakterizedir. EOG subnormal olarak saptanırken; ERG ve karanlık adaptasyon değerleri normal sınırlarda olur. En sık karşılaşılan semptom hafif düzeyde görme kaybı veya hafif metamorfopsidir. Ancak, hastalık yaşla birlikte ilerleyerek peripapiller bölgeye kadar uzanan atrofik, depigmente lezyonlara neden olabilir ve nispeten düşük olmakla birlikte ileri yaşlarda koroid neovaskülarizasyonu (KNV) riski bildirilmiştir. Her iki patoloji de santral görmeyi etkileyebilir ve dolayısıyla bu hastalığın en önemli prognostik faktörleridir.
Histopatolojik çalışmalarda koryokapillarisin korunduğu, ancak fotoreseptör hücre katmanının ve RPE'nin tamamen yok olduğu ve subretinal boşlukta hüre içi lipofuksin birikimi olduğu gösterilmiştir. RPE atrofisi dışında kalan bölgelerde lipofuksin birikimi RPE'yi belirgin olarak kalınlaştırmaktadır. Uzun bir süreçte gelişen pigment birikimi ve atrofik lezyonlar, fundus görünümünü oluşturmaktadır.
1- Kelebek Şekilli Patern Distrofi: Makülada biriken pigmentin çeşitli desenler oluşturmasıyla karakterize bir hastalıktır. RDS/periferin geninde mutasyon saptanmıştır. Otozomal dominant geçişli olup 2-3. Dekatta ortaya çıkar. Ancak otozomal resesif kalıtım şekli de bildirilmiştir. Bazı hastalarda kelebek şeklinde, diğerlerinde farklı şekillerde olabilir. Semptomlar hafif bir görme azalması şeklinde olabilir. Hastalar bazen metamorfopsi tanımlayabilirler. Çoğu hasta rutin incelemede fark edilir. Göz dibi bulguları oldukça simetrik olup bilateral, çoğu kez kelebek deseni şeklinde veya amorf pigment birikimi saptanır. Birikim RPE düzeyindedir. Yüzeyel retina katları optik disk ve damarlar normaldir. Hastalık çok yavaş ilerler. Kelebek desenli distrofide KNV gelişme riski vardır. ERG normaldir, EOG ise olguların çoğunda subnormaldir. Bazı olgularda ise vitelliform lezyon görülür, bunlarda EOG bozulmuştur. Hatta bazılarında vitelliform distrofideki bulguları hatırlatır. Histopatolojik incelemede RPE ve fotoreseptör tabakasında kayıp saptanmıştır. Normal koryokapillaris ve subretinal alanda lipofuksin dolu hücre birikimi bulunmuştur. Flöresein anjiyografide (FA) pigment birikimlerinin olduğu bölgeler hipoflöresandır, çevrelerinde ise hafif bir hiperflöresans saptanır. Fundus otoflöresans görüntülemede (FAF) lezyonun pigmentli kısımları melanolipofuksin yapısına bağlı olarak artmış otoflöresans gösterir. Depigmentasyon zonu ile çevrilmiş benekli pigment paterni mevcuttur. Çizgilerin etrafındaki depigmetasyon alanı FAF görüntüsünde azalmış otoflöresans olarak kendini göstermektedir. OCT'de biriken pigmente ait RPE elevasyonu saptanır. Görme keskinliği normal veya hafifçe azalmıştır. İleri yaşlarda atrofi ile ilişkili olarak görme kaybı artar. RPE atrofisi alanları hipo-otofloresan olarak izlenir. Ayırıcı tanıda genellikle zorluk çekilmez. Best, fundus flavimakülatus, rubella retinopatisi, toksik retinopatiler göz önünde bulundurulmalıdır.
2- Stargardt/Fundus Flavimakülatusa Benzeyen Multifokal Patern Distrofi: Sıklıkla RDS/periferin mutasyonu sonucu gelişir, otozomal dominant geçişlidir ve fenotipik olarak Stargard/Fundus flavimakülatusa oldukça benzerlik gösterir. Parafoveal pisciform lezyonlar görülür. FAF görüntülemede, arkad çevresinde ve makülada, sınırında azalmış otoflöresans olan hiperotoflöresan benekler izlenir. Hastalığın ilerleyen dönemlerinde bu benekler daha konfluent hale gelerek, halka şeklinde maküla ve optik diski çeviren hipootoflöresan yamalar oluşturur. Hastalığın başlarında halka merkezinde artmış otoflöresan izlenirken ilerleyen dönemlerde atrofiye bağlı hipootoflöresans görülür. Bu olgularda Stargardt’dan farklı olarak, peripapiller alanda da tutulum görülür. Ayırıcı tanıda; hastalığın otozomal dominant geçiş göstermesi, göreceli olarak geç yaşta başlaması, görme düzeyinin iyi olması, FAF’ta peripapiller korunmuş alanın bulunmayışı ve FA’de karanlık koroid bulgusunun olmayışı yol gösterici olabilir.
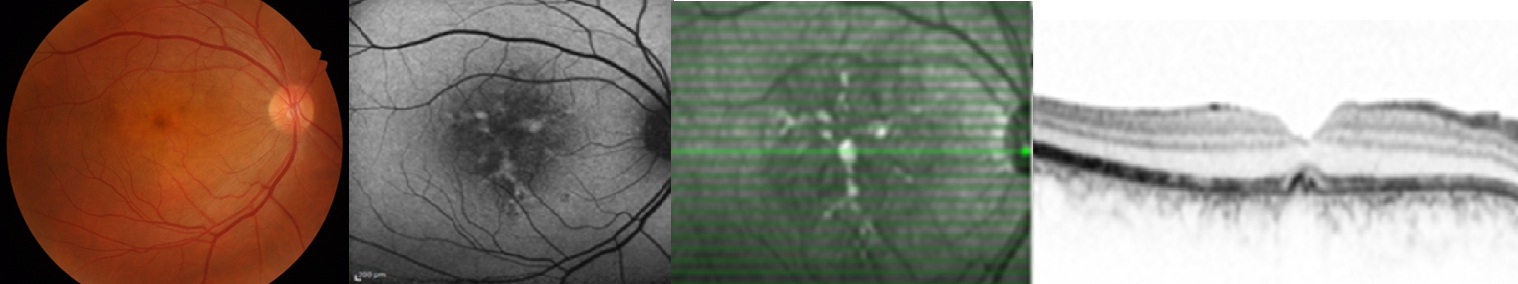
3- Retiküler Distrofi (Sjögren Distrofisi): Her iki arka kutupta maküla çevresinde ince pigment granüllerinden oluşan balık ağı şeklinde bir yapı ve foveada koyu renk pigment birikimi ile seyreden bir patern distrofidir. Otozomal dominant ve resesif geçiş söz konusudur. Vizyon genellikle normal veya normale yakındır. Çoğunlukla rutin muayenede saptanır. Başlangıçta foveada pigment birikimi oluşur, daha sonra çevresinde balık ağı şeklinde çok köşeli pigment birikimleri meydana gelir. İlerlemiş olgularda pigment dağılır ve düzensiz bir şekil alabilir. FA'da pigment birikimi olan alanlarda hipoflöresans diğer alanlarda normal koroid floresansı saptanır. Retiküler desen FAF'da çok tipiktir. Lipofuksin birikimi ve RPE atrofisi olan diğer distrofilerde olduğu gibi özellikle de RDS/periferin geni mutasyonu olanlarda pigment birikimi olan bölgelerde artmış otoflöresans bulgusu vardır. ERG normaldir, EOG de ise normalin alt sınırında değerler elde edilebilir. Yaşlılarda özellikle druzen olanlarda perifer ve midperiferde retiküler tarzda RPE değişimleri saptanabileceği unutulmamalıdır Ayrıca angioid streaks, dominant druzen ve flavimakülatus olgularında da retiküler desen görülebilir.
4- Fundus pulverulentus: Tüm patern distrofiler içinde en nadir olan türüdür. Genellikle orta-ileri yaşlarda görme azlığı ve metamorfopsi şikâyeti ile başvururlar. RDS/periferin mutasyonu sonucu gelişir, otozomal dominant geçişlidir. Maküla alanında iri ve noktasal pigment epitel beneklenmesi ve makülada granüler görünüm ile karakterizedir. Bazen bu pigmentasyon değişikliklerini diğer birçok makülopatide görülenlerden ayırt etmek zordur. ERG ve EOG normal ya da subnormal olabilir. FA’da pigment beneklenme alanına uyan bölgede hipoflöresan noktalar görülür. FAF’da pigment birikimi olan bölgelerde artmış otoflöresans bulgusu mevcuttur.
5- Erişkin başlangıçlı (Adult) foveomaküler vitelliform distrofi: Patern distrofilerin en sık görülen formudur. Genetik heterojenite gösteren polimorfik bir hastalıktır. Yapılan genetik analizlerde sıklıkla RDS geninde ya da bestrofin geninde mutasyon saptanmıştır. EBFVDde histopatolojik olarak RPE’de düzensizlik ve hiperplazi, subretinal ekstraselüler debris ya da lipofuksin birikimi gösterilmiştir.
Lezyonlar genellikle bilateral sarı renkli, bir disk çapından küçük, merkezinde pigmentasyon içeren subfoveal birikimler şeklinde kendini gösterir. Hastalar erken evrelerde görsel açıdan asemptomatiktirler veya tek ya da çift taraflı hafif görsel bulanıklık veya metamorfopsi şikâyetleri olabilir. FAF görüntülemede santraldeki birikime karşılık gelen alan hiperotoflöresandır. Bazı olgulardaki lezyonlar Best vitelliform distrofisi’ndeki evrelere benzer şekilde santraldeki birikim klasik yuvarlak yada oval şekilden lezyonun zaman içinde değişimi ile psödohipopiyon yada çırpılmış yumurta görünümüne dönebilir. Yine takiplerde skar gelişimi ile görmede düşme olabilir. Fokal birikime göre yamalı otoflöresans gösteren olguların daha düşük görme keskinliği ve retinal sensivite ile ilişkili olduğu bulunmuştur. Near-infrared (Uzun dalga boylu) otoflöresans görüntüleme, bu olguların tanısında oldukça duyarlıdır. OCT’de EBFVD'de hiperreflektans veya gölgelenme olmadan santral subretinal kalınlaşma gözlenir.
EBFVD, yaş tip maküla dejenerasyonun ayırıcı tanısında düşünülmesi gereken patolojilerden birisidir. Vitelliform lezyonlara koroid neovaskülarizasyonu düşünülerek hatalı tedaviler uygulanabilmektedir. FAF görüntüleme bu iki hastalığın ayırımında oldukça faydalı ve pratik kabul edilebilecek bilgiler vermektedir. Yaşa bağlı maküla dejenerasyonunda FA’de KNV sızıntısına bağlı hiperflöresans mevcut iken, EBFVD'de de lezyonun boyanmasına bağlı aynı şekilde hiperflöresans izlenir. Fakat FAF’da KNV blokaja bağlı olarak hipootoflöresan görüntü varken, vitelliform lezyonda hiperotoflöresans görülür. Bunun yanı sıra KNV patern distrofiler içinde en sık (%5-15) EBFVD ile birlikte bildirilmiştir
Sonuç: PD'ler RPE'nin benign distrofileridir ve prognozları KNV gelişmedikçe iyi olma eğilimindedir. PD'ler KNV'nin etiyolojik ayırıcı tanısında unutulmaması gereğidir. Druzen ve diffüz skar yokluğu PD'yi YBMD’yi ayırt eden oldukça karakteristik bulgulardır. KNV, PD'de santral görme kaybına yol açan en önemli komplikasyon kabul edilmektedir. KNV'nin distrofinin direkt sonucu mu olduğu yoksa distrofinin yalnızca bu gözleri KNV gelişimine yatkın hale mi getirdiği bilinmemektedir. Tüm PD alt tiplerinde KNV gelişme riski düşük olmakla birlikte vardır. KNV gelişen olgularda prognoz YBMD'de olduğu kadar kötü gözükmemektedir. Ayrıca, idiyopatik olduğu kabul edilen KNV'lerin etiyolojik ayırıcı tanısında PD her zaman akılda tutulmalıdır. Bazı idiyopatik olduğu kabul edilen KNV olgularında, KNV’lerin altta yatan PD bulgularını kapatabileceği ve PD ile olan ilişkinin tanınmayabileceği akılda tutulmalıdır. Ayrıca, bazı olgularda PD'nin tipik pigment birikintileri, idiyopatik olduğu kabul edilen KNV'den birkaç yıl sonra ortaya çıkabilmektedir. Her ne kadar PD'ye bağlı gelişen gizli KNV bildirilmişse de, olguların çoğu klasik KNV’dir ve Anti-VEGF tedaviyle iyi yanıtlar alınmaktadır.
[Kaynaklar:1- Demirel S, Batıoğlu F, Özmert E. Maküla Distrofilerinde Fundus Otoflöresans Görüntüleme. Ret-Vit 2012; 20: 250-259. 2- Özdek Ş. Hondur A, Gürelik G, Aydın B, Hasanreisoğlu B. Retina Pigment Epitelinin Patern Distrofisi ve Koroidal Neovaskülarizasyon. Ret-Vit 2006; 14: 11-16. 3- Kır N. Best Hastalığı Dışındaki Maküla Distrofileri. Turkiye Klinikleri J Ophthalmol-Special Topics 2009; 2(1): 32-8.]
Not: Web sitemizdeki bu bilgiyi, sunu ve yayınlarınızda aşağıdaki şekilde kaynak göstererek kullanabilirsiniz.
(Çıtırık M, Teke MY. Patern Distrofiler. http://www.retinaclub.com/ Son Güncellenme Tarihi 01/03/2016).